Private Label, White Label, Wholesale partnerships available - EU, USA and UK - Free shipping from €75
Water for Injection: A Guide to Purity and Safe Use
A procurement manager opens a specification sheet and sees three words that look deceptively simple: water for injection. A junior researcher reaches for a vial labelled sterile water and assumes any “clean” water will do. Then a batch fails, a reconstitution behaves oddly, or a quality review asks a harder question than expected: what exactly was in the water, and how was that risk controlled?
That's where confusion usually starts. Water looks like the most ordinary material in a lab or production setting, but in pharmaceutical work it often carries the heaviest burden. It can dissolve an active ingredient, contact a sterile pathway, influence conductivity, introduce organics, or bring in endotoxins that no one can see at the bench. In the wrong context, the water itself becomes the variable.
For that reason, Water for Injection (WFI) isn't just a “cleaner” grade of water. It's a tightly controlled material used where hidden contamination creates outsized risk. For researchers, that means protecting reproducibility and sample integrity. For buyers and distributors, that means treating WFI as a quality and continuity decision, not a commodity line item.
Table of Contents
- The Hidden Risks of Impure Laboratory Water
- Defining Water for Injection Beyond Sterility
- Decoding Pharmacopoeia Standards for WFI
- How WFI Is Manufactured and Tested for Purity
- WFI vs Bacteriostatic and Sterile Water
- Safe Handling and Application in Research
- Procurement and Compliance for Distributors
The Hidden Risks of Impure Laboratory Water
A lab can do almost everything right and still get a bad outcome because of the water. Reagents may be within expiry, glassware may be clean, the method may be sound, and yet the result still drifts. That usually happens because water is treated as a background material instead of a controlled input.
In routine bench work, lower-grade water may seem harmless. It looks clear. It may even test acceptably for a less demanding purpose. But if that same water carries trace ions, residual organics, viable microbes, or endotoxins, it can subtly interfere with reconstitution, analytical performance, or a sterile manufacturing step. The problem is that these failures rarely announce themselves clearly. They show up as inconsistency.
A useful way to think about it is this. If a formulation is like a clean surgical tray, the water is not just the tray liner. It touches everything on the tray. Any impurity it carries can travel into the final preparation.
Impure water doesn't have to look dirty to create risk. In pharmaceutical work, the most serious contaminants are often invisible.
That's why Water for Injection sits at the top of the laboratory and pharmaceutical water hierarchy for critical uses. It exists for situations where ordinary purity isn't enough, and where the consequences of microbial fragments or chemical residues are too serious to ignore.
A common point of confusion
Many new buyers assume that “sterile water” and WFI mean the same thing. Many junior researchers assume the difference is mostly regulatory wording. Neither assumption is safe.
The key issue isn't only whether living microbes are absent. It's whether the water has been controlled tightly enough for applications where pyrogens, ionic contamination, and organic carryover all matter. In that sense, WFI is less about prestige and more about risk prevention.
For procurement managers, that changes the purchase question from “Which water is available?” to “Which water grade fits the application, and what failure does it prevent?” For researchers, it changes the handling question from “Is the vial sealed?” to “Does the water still meet the integrity needed for this experiment?”
Defining Water for Injection Beyond Sterility
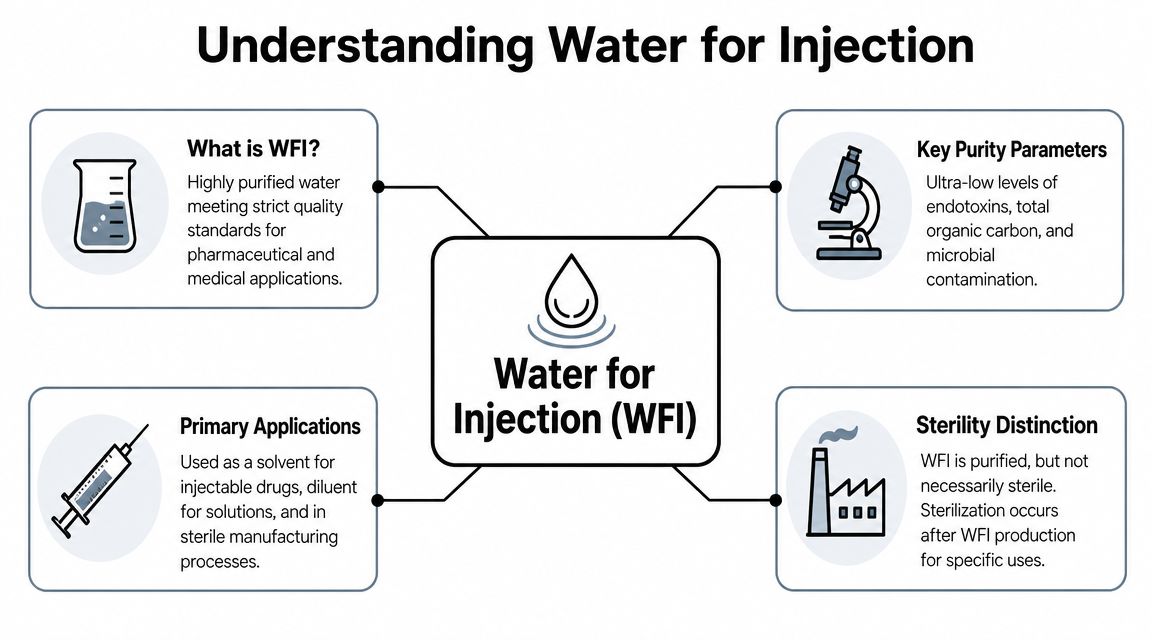
Many people first understand WFI as “very pure sterile water”. That's a useful starting point, but it's incomplete. Water for Injection is defined by a stricter purity objective than sterility alone.
Sterile is only part of the story
Sterile means living microorganisms have been removed or destroyed. That matters, but it doesn't answer a second question that is just as important in injectable and high-risk applications: what about the non-living residues those microorganisms can leave behind?
The most important example is endotoxin. Endotoxins are fever-causing components associated with certain bacteria. A sample can be free of live bacteria and still contain endotoxin. That's why people who are new to WFI often misunderstand the target. The goal isn't only “nothing alive”. The goal is also “nothing left behind that can trigger harm”.
An analogy helps. A normal clean room protects against obvious dirt and active contamination. A microchip fabrication space controls even finer invisible particles because the process can't tolerate them. WFI works in a similar way. It isn't just water that looks clean or even microbiologically quiet. It's water controlled to a level that makes it suitable for preparing injectable medicines and other highly sensitive materials.
What WFI is really designed to control
WFI is best understood by what it lacks:
- Very low ionic contamination so dissolved salts and charged impurities don't disturb the final preparation.
- Very low organic contamination so traces of organic material don't interfere with stability, analysis, or process performance.
- Tight microbial control so viable contamination remains extremely limited.
- Tight endotoxin control so pyrogenic risk stays low in applications linked to parenteral use.
That last point is the one that often separates basic understanding from competent decision-making. A researcher may only think about whether a powder dissolves. A quality team thinks further ahead. Could this water introduce a variable that changes cell response, assay performance, or a sterile process? Could it compromise a preparation intended for injection-related use? WFI exists to reduce those possibilities.
A second area of confusion involves the word “injection” itself. Some readers assume that means WFI is only relevant inside finished injectable drug products. In practice, its importance extends into manufacturing support, solution preparation, equipment-related uses, and research activities where a very high-purity aqueous vehicle is needed.
Practical rule: If the application is sensitive enough that unseen residues could distort the outcome or create safety concerns, “clean enough” is not a meaningful standard.
For new procurement staff, this means the label should never be read in isolation. The grade, intended use, documentation, and handling conditions matter together. For junior researchers, it means a vial of WFI should be treated less like a convenience supply and more like a controlled reagent.
Decoding Pharmacopoeia Standards for WFI
Specifications are where WFI stops being a general idea and becomes a measurable standard. For procurement teams and QA staff, this is the part that turns “high purity” into something that can be verified.
According to European Pharmacopoeia-aligned WFI specifications described by Veolia Water Technologies, conductivity must be below 1.1 μS/cm at 20°C, total organic carbon must be ≤0.5 mg/L, bacterial count must be under 10 cfu/100 mL, and endotoxins must be below 0.25 IU/mL. Those aren't abstract numbers. Each one targets a different contamination risk.
What the main limits mean in practice
Conductivity is a fast way to assess ionic contamination. If conductivity rises, dissolved charged species are usually present. In plain terms, that means salts or other ionisable contaminants may have entered the water. In formulation and analytical work, those ions can alter solution behaviour, disturb sensitive systems, or point to broader control problems upstream.
Total Organic Carbon (TOC) tracks organic residues. This doesn't identify every molecule individually. Instead, it asks a broader question: how much organic material is present overall? A high TOC result can suggest contamination from feedwater carryover, materials in contact with the system, or cleaning-related residues. For any process that relies on a controlled solvent background, that matters.
Bacterial count addresses viable microorganisms. This is straightforward but still easy to oversimplify. A low count doesn't only protect the final use. It also indicates that the production and distribution system has remained under control. If bacterial levels rise, the water system may already be drifting out of a healthy operating state.
Endotoxins are the most misunderstood parameter for non-specialists. A low endotoxin limit exists because dead bacteria can still be dangerous in parenteral contexts. A water system that looks acceptable microbiologically can still fail on pyrogen control.
Why procurement teams should care about each line
A certificate that says “meets spec” is only useful if the buyer understands what each specification is protecting against.
| Parameter | What it indicates | Why it matters |
|---|---|---|
| Conductivity | Dissolved ionic impurities | Helps protect against unintended chemical interference |
| TOC | Organic contamination | Helps control residue-related variability |
| Bacterial count | Viable microbial burden | Reflects microbiological control of the system |
| Endotoxins | Pyrogenic contamination | Critical for injection-related and highly sensitive uses |
For a junior researcher, these values explain why one water grade performs predictably while another introduces noise. For a procurement manager, they define what should be checked on batch documentation and what questions should be raised when results trend close to limits.
The specification sheet is not administrative paperwork. It is a map of the risks the water has been designed to suppress.
That's also why WFI should never be chosen by name alone. The meaningful question is whether the supplied material demonstrably meets the required pharmacopoeial standard, with documentation that supports release, traceability, and intended use.
How WFI Is Manufactured and Tested for Purity
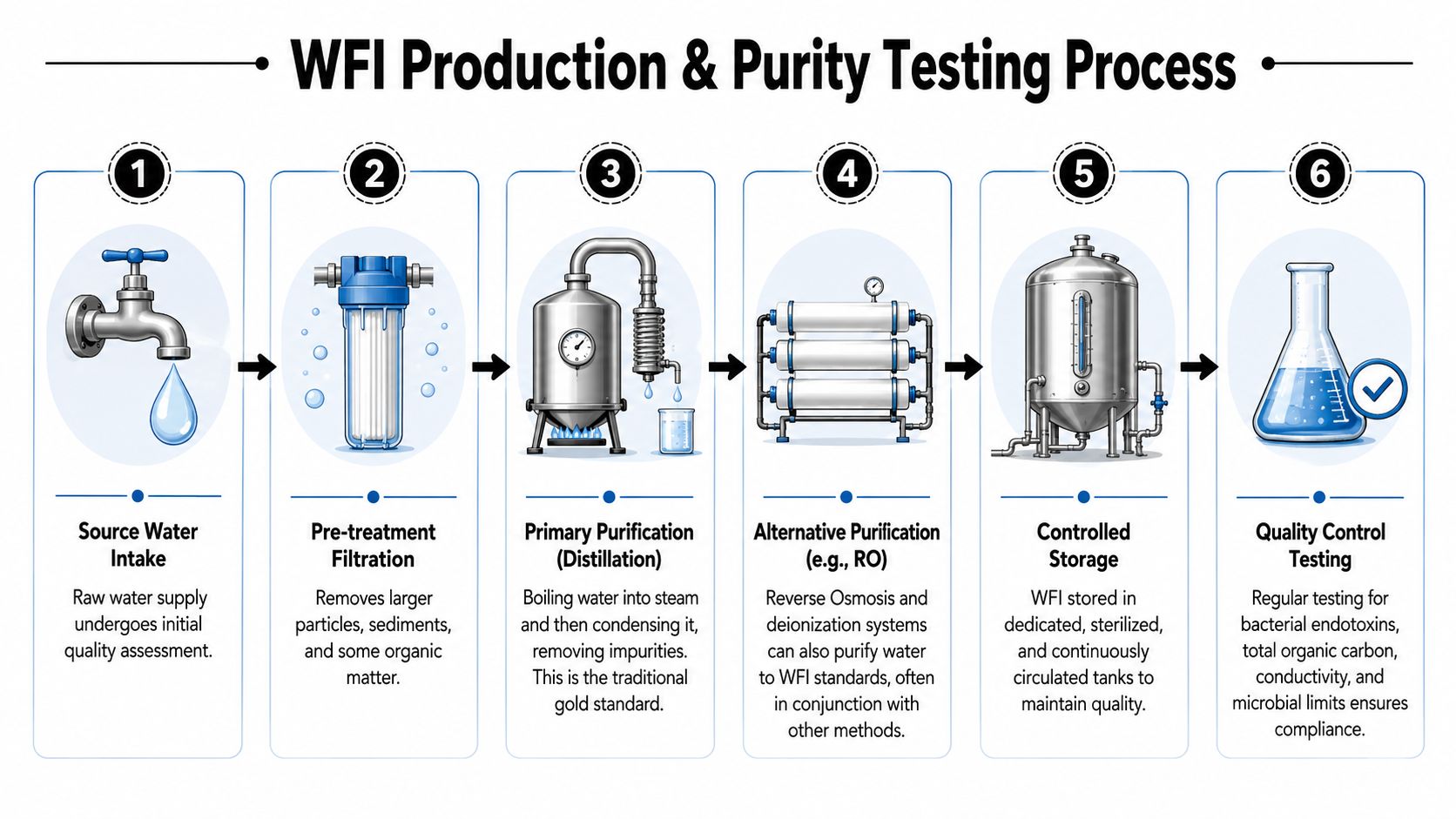
The purity level required for WFI doesn't come from a single filter or a single pass through a machine. It comes from a controlled sequence of purification, storage, circulation, and testing. Each part exists because contaminants behave differently. Some are dissolved ions. Some are organics. Some are microorganisms. Some are endotoxins. No single step handles all of them equally well.
Early in the process, it helps to see the whole path from raw water to final release.
Two accepted routes to the same purity goal
A key regulatory milestone came in the EU in 2017, when the European Pharmacopoeia revised its monograph so that WFI became formally available as a membrane-produced bulk water grade, including technologies such as reverse osmosis, while maintaining stringent purity controls, as outlined in Cytiva's explanation of the EU WFI monograph change. That change matters because it clarified that distillation is no longer the only accepted route in the EU.
Today, accepted production routes include:
- Multiple-effect distillation
- Vapour compression distillation
- Membrane-based systems
These routes serve the same purpose through different engineering logic. Distillation separates water from many impurities by evaporation and condensation. Membrane systems rely on stages such as reverse osmosis and related polishing steps to remove dissolved ions and microbiological contaminants. In both cases, the process isn't judged by tradition. It's judged by whether the output meets the required WFI standard consistently.
For readers who work around material quality more broadly, material purity in experimental integrity is a useful parallel. The same principle applies here. The less controlled the input, the more hidden variability enters the result.
Why storage and circulation matter as much as generation
Producing clean water is only half the challenge. Keeping it clean is the harder part.
According to MECO's guide to Water for Injection generation, WFI production in the IE-region pharmaceutical supply chain is typically validated as a continuously circulating, high-temperature utility above 80°C, because heat suppresses microbial proliferation. The same source explains why pretreatment such as reverse osmosis is used to remove dissolved ions, bacteria, viruses, and suspended solids that would otherwise increase conductivity, promote scale, or corrode distillation equipment.
That detail is easy to overlook, but operationally it's vital. A beautifully designed generation system can still fail if storage tanks, loops, valves, or sampling practices allow regrowth or recontamination.
A short technical video can help readers visualise how these systems fit together in practice.
Testing turns engineering into evidence
Even the best-designed system needs routine proof. In practice, suppliers test WFI to confirm that the final water remains within required limits for conductivity, TOC, microbiological quality, and endotoxins. That testing is what converts process design into batch confidence.
A useful way to frame it is this:
- Pretreatment reduces the burden on the main purification train.
- Primary purification removes major classes of contaminants.
- Storage and circulation preserve the state achieved.
- Testing verifies that the system delivered what it was meant to deliver.
A WFI system is not just equipment. It is an ongoing control strategy.
For buyers, this is why manufacturing method and test documentation belong in the same conversation. For researchers, it explains why a vial of WFI carries more behind it than a label and a fill volume.
WFI vs Bacteriostatic and Sterile Water
Practical mistakes commonly occur. A researcher needs a diluent quickly. A reseller lists several water products that sound similar. Someone sees “sterile”, someone else sees “for injection”, and the distinction gets flattened into a naming issue. It isn't one.
The comparison that prevents expensive mistakes
The biggest divider is the presence or absence of a bacteriostatic agent. Bacteriostatic water contains an antimicrobial preservative. Sterile water for injection does not. WFI, in its core sense, refers to the highly purified water grade defined by pharmacopoeial requirements and used where that stricter purity profile is needed.
That means the choice shouldn't start with convenience. It should start with intended use, preservative tolerance, and whether the application can accept anything other than highly purified water.
For a more detailed product-level overview, this guide to bacteriostatic and sterile water is useful when teams need a practical reference for reconstitution decisions.
Comparison of Laboratory Water Types
| Attribute | Water for Injection (WFI) | Bacteriostatic Water | Sterile Water for Injection |
|---|---|---|---|
| Core composition | Highly purified water meeting WFI standard | Sterile water with a bacteriostatic preservative | Sterile water without a bacteriostatic preservative |
| Preservative present | No | Yes | No |
| Main decision factor | Highest purity for critical applications | Multi-use handling context where preservative is intended | Sterile diluent use without preservative |
| Endotoxin focus | Explicitly critical | Product-specific documentation must be checked | Product-specific documentation must be checked |
| Typical concern | Protecting against ionic, organic, microbial, and pyrogenic risk | Avoiding misuse where preservative is unsuitable | Avoiding confusion with WFI-grade bulk water |
| Use choice | Driven by purity requirement | Driven by preservative-based use case | Driven by sterile single-use style applications |
How to choose the right option for the task
Some quick decision rules help.
- For injection-related or highly sensitive pharmaceutical use, WFI is the reference point because the purity target goes beyond simple sterility.
- For reconstitution where a preservative would interfere, bacteriostatic water may be the wrong choice even if it seems convenient.
- For routine sterile dilution, sterile water may be appropriate only when the application and documentation support it.
- For repeated vial access, teams need to understand whether the product was designed for that pattern of use or whether each entry increases contamination risk.
One of the most common errors is assuming bacteriostatic water is “better” because it resists bacterial growth after opening. That reasoning ignores compatibility. A preservative may be useful in one context and completely unsuitable in another.
If the additive changes the chemistry or the permitted use, the wrong water can fail even when it looks more practical on paper.
Another common error is treating WFI as a marketing phrase rather than a grade with technical meaning. Procurement teams should read the product name, composition, intended use statement, and supporting documents together. Researchers should check whether the water choice matches the protocol, not just the cap colour or vial format.
Safe Handling and Application in Research
Even the right water grade can be compromised by poor handling. Once a container is opened, the question changes. It's no longer only about how pure the product was at release. It's about whether the user preserves that condition long enough to complete the work.

Where high-purity water helps most
In research settings, WFI or WFI-style sterile diluents are often chosen when the water will directly contact a sensitive material.
Examples include:
- Peptide reconstitution where dissolved impurities can affect solubility or introduce experimental noise.
- Preparation of certain cell-related solutions where background contamination may alter behaviour in subtle ways.
- High-purity analytical workflows where unwanted ions or organics can show up as interference rather than visible contamination.
In these contexts, the water is part of the method. It shouldn't be treated as a generic consumable.
One supplier option in this broader category is Herbilabs, which offers sterile reconstitution solutions in premium glass vials for research use only. The relevant point for buyers isn't branding. It's that the packaging format, documentation, and stated use should match the workflow the lab is running.
Handling habits that protect the water after opening
Aseptic discipline matters here. Essential aseptic techniques for accurate lab research gives a useful operational reference, but the core habits are straightforward:
- Disinfect the closure before access. A clean stopper reduces the chance of carrying contamination into the vial.
- Use sterile, suitable withdrawal tools. A contaminated needle or tip defeats the purpose of starting with high-purity water.
- Minimise open time. The longer the container remains exposed, the greater the chance of environmental contamination.
- Label after opening. If a lab permits temporary post-opening storage for a defined workflow, staff should record identity and opening details clearly.
- Follow the product's handling instructions. Storage and shelf-life decisions should come from the product documentation and the lab's SOPs, not habit.
A junior researcher often focuses on getting the powder into solution. A senior scientist looks one step earlier and one step later. Was the water withdrawn aseptically, and was the solution used in a way that preserved its intended quality?
The vial doesn't stay “as supplied” once handling begins. The user becomes part of the control system.
That mindset improves day-to-day work immediately. It reduces accidental contamination, improves reproducibility, and helps labs defend their methods when questions arise later from supervisors, collaborators, or auditors.
Procurement and Compliance for Distributors
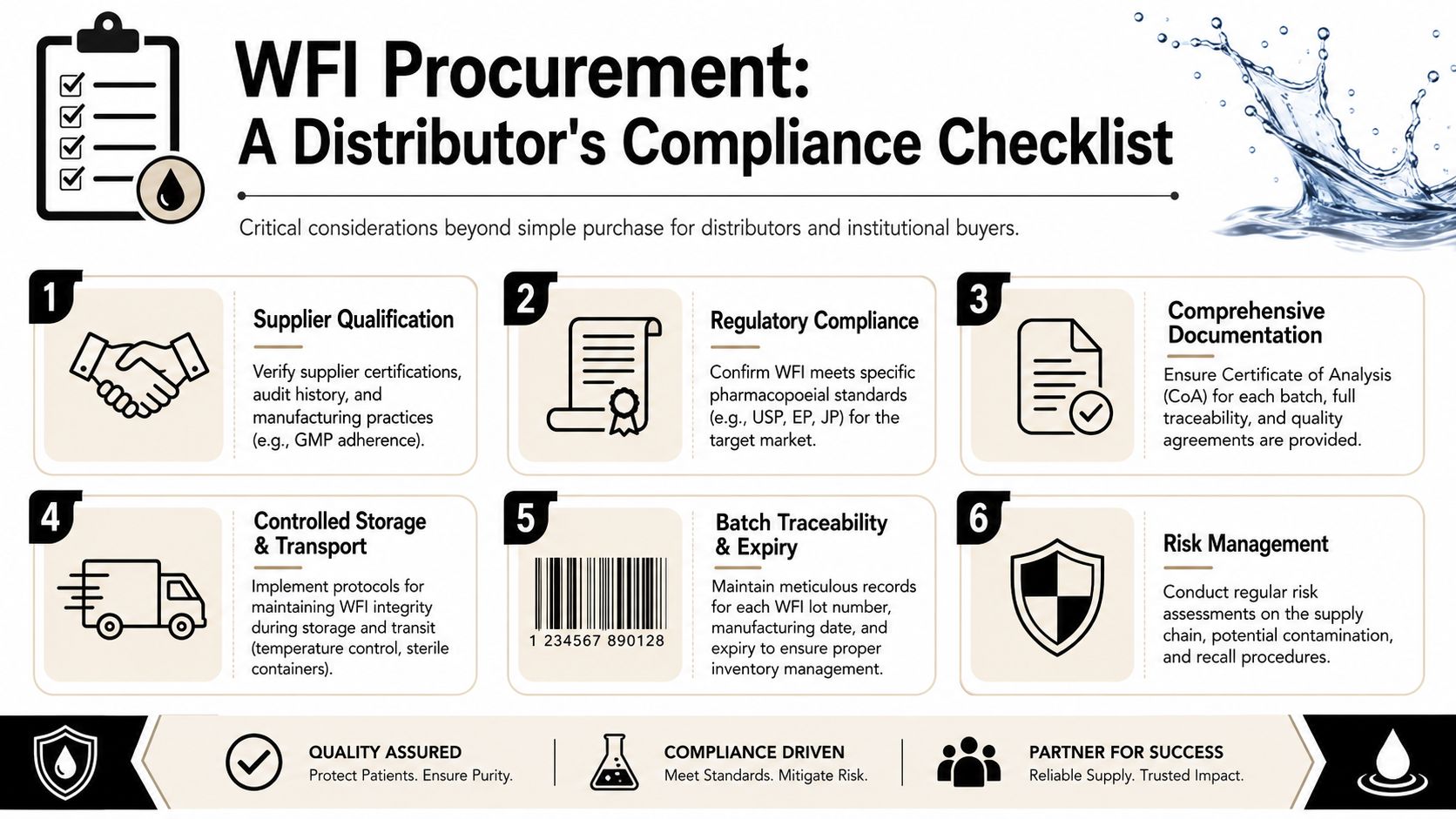
For distributors and institutional buyers, WFI procurement is not a simple exercise in matching item codes to demand. It sits inside a broader quality system. The product, the batch record, the transport conditions, the traceability chain, and the intended market status all matter together.
Documentation is part of the product
A distributor should expect more than a label claim. Batch-specific documentation, especially a Certificate of Analysis, supports verification that the material supplied matches the required standard. That document helps downstream customers confirm compliance with relevant specifications and gives the distributor something concrete to retain for traceability and complaint handling.
The practical checklist usually includes:
- Batch documentation that matches the shipped lot
- Traceability records linking receipt, storage, and onward sale
- Clear intended use statements, especially where products are sold for research use only
- Storage and transport controls that preserve integrity through fulfilment
A buyer who treats documentation as overhead usually creates more risk later. When a customer questions a lot, asks for supporting evidence, or needs rapid trace-back, the distributor with complete records moves faster and with less exposure.
Supply resilience needs active planning
Growth in the market doesn't remove fragility from the supply chain. According to Supply Chain Dive's report on sterile water shortages and WFI market projections, sterile water for injections was reported in shortage starting in December 2017, shortages were still described as widespread at the time of the report, and one 2026 projection expects the global WFI market to grow from USD 35.72 billion in 2026 to USD 62.82 billion by 2033, with an 8.4% CAGR and North America projected to hold 42.3% of the market in 2026. The operational lesson is simple. Rising demand can increase dependence on validated capacity and dependable distribution rather than make procurement easier.
For distributors, that points to a few sensible priorities:
- Qualify more than one route where possible. Single-source dependence can become a continuity problem.
- Review supplier capability, not just availability. A supplier should be able to support documentation and stable fulfilment.
- Build internal recall readiness. Lot-level control matters before a problem occurs, not after.
- Keep RUO boundaries clear. Product positioning, listing language, and customer communication should align with the permitted use.
Buyers don't just purchase WFI. They purchase the supplier's ability to keep quality intact from release to receipt.
That's what separates low-friction distribution from avoidable compliance stress.
Herbilabs supports research-focused buyers with sterile diluents, reconstitution solutions, batch documentation, and wholesale supply options across multiple markets. Teams reviewing suppliers can explore Herbilabs for product information, compliance context, and distribution partnership enquiries.
Published via Outrank


